면역글로불린 G4 연관질환의 개요
Overview of the Immunoglobulin G4-related Disease Spectrum
Article information
Trans Abstract
Immunoglobulin G4 (IgG4)-related disease is a newly named fibroinflammatory condition characterized by tumefactive lesions that contain dense lymphoplasmacytic infiltrates rich in IgG4-positive cells. Elevated serum IgG4 concentrations also often accompany IgG4-related disease. Many medical conditions that were long viewed as diseases confined to single organs (e.g., Mikulicz disease, type 1 autoimmune pancreatitis, Riedel’s thyroiditis, multifocal fibrosclerosis, inflammatory pseudotumor, mediastinal fibrosis, retroperitoneal fibrosis, and etc.) are now designated as part of the spectrum of IgG4-related disease. The preferred nomenclature suggested by a committee of international experts uses the prefix “IgG4-related-” for individual organ involvement, regardless of the organ system affected. One exception is type 1 autoimmune pancreatitis (IgG4-related pancreatitis). Comprehensive diagnostic criteria for IgG4-related disease and organ-specific diagnostic criteria (e.g., IgG4-related dacryoadenitis and sialadenitis, type 1 autoimmune pancreatitis, IgG4-related kidney disease and IgG4-related sclerosing cholangitis) can aid clinicians in the diagnosis of this erratic condition.
INTRODUCTION
Immunoglobulin G4 (IgG4)-related disease is a newly named multi-organ fibroinflammatory condition characterized by tumefactive lesions that contain dense lymphoplasmacytic infiltrates rich in IgG4-positive cells [1-3]. It is often accompanied by elevated serum IgG4 concentrations. The concept of IgG4-related disease was recognized after 2003, with the identification of various extrapancreatic manifestations of autoimmune pancreatitis (AIP) [4]. Subsequent research has revealed that IgG4-related disease can involve virtually every organ system: the pancreas, biliary system, salivary glands, kidneys, retroperitoneum, liver, gallbladder, thyroid, lungs, aorta, prostate, stomach, periorbital tissues and pericardium [1]. Awareness of IgG4-related disease is relevant to clinicians because this disorder can dramatically respond to glucocorticoids [3,5]. The aims of this review are to introduce the newly recognized concept of IgG4-related disease and to address the spectrum and nomenclature of this disorder.
DISEASE CONCEPT
The concept of type 1 AIP, referred to as IgG4-related pancreatitis, was established before the concept of IgG4-related disease emerged. Type 1 AIP represents a unique subset of chronic pancreatitis with various extrapancreatic manifestations including bile duct, salivary gland, thyroid, and kidney involvement [1]. IgG4-related disease of the biliary tree, referred to as IgG4-related sclerosing cholangitis (IgG4-SC), typically presents with accompanying AIP. Pancreatic and extrapancreatic tissue of patients with AIP showed strikingly similar histopathological features, including dense lymphoplasmacytic infiltrates rich in IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis. Based on these findings, Kamisawa et al. [4] suggested the new clinicopathological entity of IgG4-related autoimmune disease. Since the systemic involvement of IgG4-related disease was first reported, many medical conditions that were long viewed as diseases confined to single organs are now designated to the part of the spectrum of IgG4-related disease (Table 1) [1]. These are Mikulicz disease, Küttner’s tumor, Riedel’s thyroiditis, multifocal fibrosclerosis, inflammatory pseudotumor (affecting the orbit, lungs, kidneys, and other organs), mediastinal fibrosis, retroperitoneal fibrosis, inflammatory aortic aneurysm, and idiopathic hypocomplementemic tubulointerstitial nephritis with extensive tubulointerstitial deposits [1].
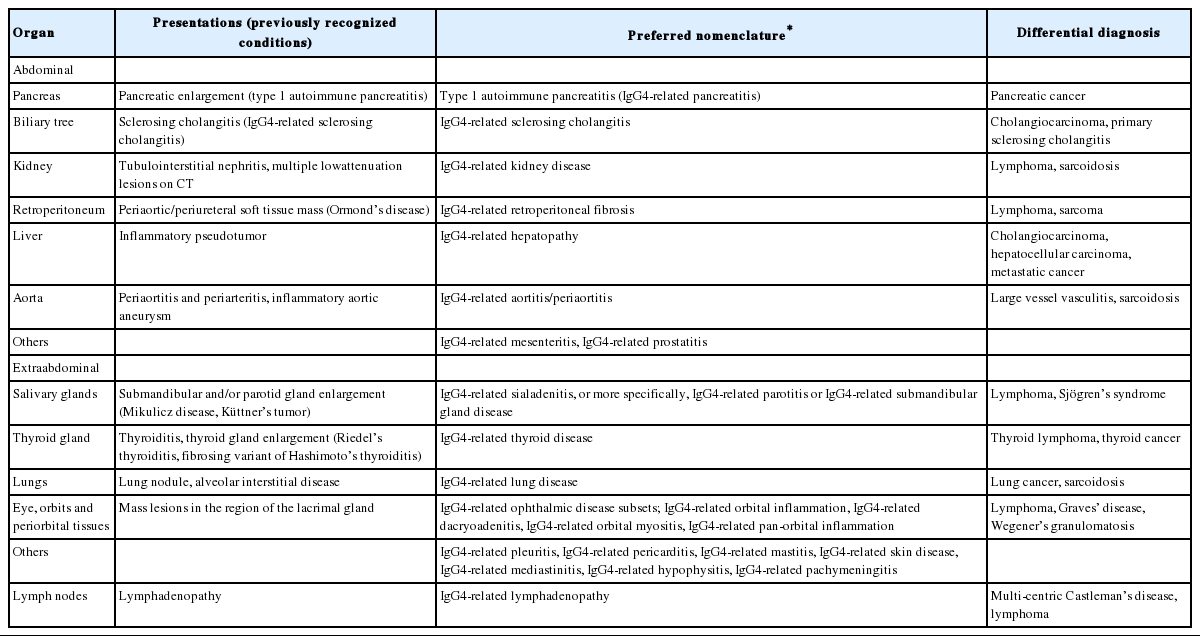
Spectrum of IgG4-related disease and preferred nomenclature
This disorder has had many different names: IgG4-related disease, IgG4-associated disease, IgG4-related systemic disease, IgG4-related sclerosing disease, IgG4-related systemic sclerosing disease, IgG4-related autoimmune disease, Hyper-IgG4 disease, IgG4-positive multiorgan lymphoplroliferative syndrome, systemic IgG4-related plasmacytic syndrome, and IgG4-syndrome [6]. An agreement on the name “IgG4-related disease” was recently reached for this multifocal disorder by Japanese investigators, and since then has found worldwide use [2,6-7].
Inclusion of IgG4 in the terminology of the disease should not lead clinicians to make a diagnosis solely based on elevated serum IgG4 or on increased tissue IgG4-positive plasma cell infiltration [6]. Rather, the diagnosis of IgG4-related disease should continue to be made by a combination of clinical, serological, and histopathological examinations, including light microscopic features and IgG4 staining [6,7]. Stone et al. [6] suggested that IgG4-related disease is analogous to sarcoidosis, another systemic disease in which diverse organ manifestations are linked by the same histopathological characteristics [1].
1. The IgG4 molecules
IgG4 is the least abundant IgG subclass, accounting for less than 5% of total IgG in healthy individuals [1]. A unique characteristic of IgG4 is its fragment antigen-binding (Fab)-arm exchange, which means that an exchange can occur where half a molecule (heavy chain-light chain pair) from one IgG4 is swapped for half of a different IgG4 antibody [1,8]. The IgG4 molecule can therefore have two distinct Fab arms, each with a different epitope specificity, thereby becoming bispecific, but remaining functionally monovalent [1,8]. These monovalent IgG4 molecules are unable to crosslink antigens, leading to ineffective immune-complex formation [1]. Another unique property of IgG4 is the fragment crystallizable (Fc)-Fc binding, which is the capacity to bind to other antibodies of IgG subclasses via interactions between their Fc portions [8]. Fc-Fc binding could potentially prevent immune responses by blocking interactions with complement proteins and Fc receptors. At present, the unanswered question is whether IgG4 antibodies are pathogenic tissue-destructive immunoglobulins or are signs of reactive overexpression in response to unknown primary inflammatory stimuli due to their anti-inflammatory properties [1,8]. The cited articles provides further explanation with illustrations [1,8].
2. Pathologic consensus statement
In parallel with the consensus diagnostic criteria proposed by internist societies, pathologists have published a consensus statement on the pathology of IgG4-related disease [9]. They acknowledged that the histological features associated with IgG4-related disease (dense lymphoplasmacytic infiltrate, fibrosis, arranged at least focally in a storiform pattern and obliterative phlebitis) are highly specific when viewed in conjunction with an IgG4 immunostain. Based on this premise, a 3-tiered diagnostic terminology for the pathological diagnosis of IgG4-related disease was endorsed: (1) histologically highly suggestive of IgG4-related disease, (2) probable histological features of IgG4-related disease, and (3) insufficient histopathological evidence of IgG4-related disease [9]. A full discussion of this issue is beyond the scope of this article.
Abundant IgG4-positive plasma cell infiltration at involved tissue sites is a disease hallmark, even in cases with normal serum IgG4 [3,9]. The cutoff for the number of IgG4-positive plasma cells in a biopsy or surgical specimen has also been proposed by international consensus [9]. The consensus statement asserts that the appropriate cutoff point may vary from organ to organ: 1) more than 10 IgG4-positive plasma cells per high-power field (HPF) for pancreatic biopsy, bile duct biopsy, and liver biopsy specimens; 2) more than 50 IgG4-positive plasma cells/HPF for surgical specimens of the pancreas, bile duct, and liver; and 3) more than 100 IgG4-positive plasma cells/HPF for a lymph node, salivary gland, and lacrimal gland [9]. The high numbers of IgG4-positive plasma cells should be accompanied by a ratio of IgG4 to IgG-positive plasma cells of at least 40% for the diagnosis of IgG4-related disease [3,7,9].
DISEASE SPECTRUM AND PREFERRED NOMENCLATURE
IgG4-related disease generally affects middle-aged to elderly men, rather than young women [2]. The disorder usually presents in a subacute manner and its clinical symptoms can be relatively mild because the disorder is characterized by swollen but painless organs [8]. Fatigue and weight loss of 5-10 kg can occur over months, whereas patients with IgG4-related disease rarely present with fever [3,8]. The diagnosis of IgG4-related disease can, therefore, take several months or even years after vague manifestation. This disease is often identified incidentally through radiologic findings or unexpectedly in pathological specimens [1]. Specific recommendations for virtually every IgG4-related organ system nomenclature were proposed by an organizing committee comprising international experts of clinicians, pathologists, radiologists, and basic scientists (Table 1) [6]. The recommended preferred nomenclature employs “IgG4-related-” as a prefix for individual organ involvement, regardless of the organ system affected [6]. For example, Mikulicz disease was positioned as IgG4-related dacryoadenitis and sialadenitis. Pancreatic manifestation, however, has retained the designation “type 1 AIP (IgG4-related pancreatitis)” because type 1 AIP is a widely accepted designation among gastroenterologists and pancreatic surgeons [6,8]. The term “IgG4-related pancreatitis” is added in parentheses for the purpose of educating the broader medical community about the relationship between IgG4-related disease and this subset of pancreatic disease [6].
DIAGNOSTIC CRITERIA
The current comprehensive diagnostic criteria for IgG4-related disease are the diagnostic criteria for IgG4-related disease first proposed by a consensus of Japanese experts in 2011 (Table 2) [7]. Comprehensive diagnostic criteria for IgG4-related disease may be significant in the sense that IgG4-related disease is an entity of systemic disease that involves multiple organs and one diagnostic criterion may be ideal for one disease entity. However, comprehensive diagnostic criteria for IgG4-related disease have been proposed for practical use by non-specialists [3,7]. Actually, the comprehensive diagnostic criteria for IgG4-related disease showed relatively low sensitivity in patients with type 1 AIP, mainly due to difficulties in performing pancreatic biopsy [7-8]. In cases with suspected IgG4-related disease and difficult biopsies, organ specific diagnostic criteria could then be applied. In actual practice by expert gastroenterologists, the diagnosis of IgG4-related disease of the pancreatobiliary system relies more on organ specific criteria for AIP or IgG4-SC. At present, organ specific diagnostic criteria are available for IgG4-related dacryoadenitis and sialadenitis (Mikulicz disease), type 1 AIP, IgG4-related kidney disease, and IgG4-SC [10-15]. Organ specific diagnostic criteria can be applied before, in place of, or after the use of comprehensive diagnostic criteria for IgG4-related disease [8].

TREATMENT AND RELAPSE
When discussing treatment of IgG4-related disease, it is important to determine the indication of treatment. When vital organs are involved with presenting symptoms (i.e., AIP with jaundice), aggressive treatment may be needed [1]. However, watchful waiting might be sufficient for asymptomatic involvement of non-vital organs such as indolent lymphadenopathy. Glucocorticoids are the classic first line of medical therapy [1,3]. Starting dose of prednisolone is usually 0.6-1.0 mg/kg daily [12,16]. After 2-4 weeks, the prednisolone is tapered by 5 mg every 1-2 weeks according to individual clinical setting [3]. To prevent relapse, maintenance therapy with low dose prednisolone (2.5-5 mg/day) for up to 3 years is recommended by Japanese investigators [16].
Although majority of patients with IgG4-related disease respond to glucocorticoid therapy, they commonly experience relapse of disease [1,16]. Most patients with disease relapse respond to a course of glucocorticoid therapy with similar remission rates achieved at the time of initial disease presentation [17-18]. For patients who relapse, steroids are often given at a higher induction dose which is tapered more gradually, and maintenance therapy is often more prolonged [19-20]. Azathioprine, mycophenolate mofetil, and methotrexate can be used for glucocorticoid-sparing effect or achieving additional immunosuppression in relapsed patients [1,3,18]. B cell depleting agent rituximab can also be an alternative for patients with recurrent or refractory disease [18]. Although patients with IgG4-related disease was reported to be at potentially high risk for cancer, long-term survival remains incompletely defined due to lack of long-term follow-up and preponderance in elderly population [21-22].
CONCLUSION
The concept of IgG4-related disease unified a variety of medical diagnoses previously regarded as being confined to single organ systems [1,6]. Swollen, but painless, organs are a common manifestation of this disorder. The awareness of IgG4-related disease is clinically relevant because this disorder can masquerade as malignancy, despite the relatively mild clinical symptoms of this disorder. Comprehensive diagnostic criteria for IgG4-related disease and organ specific diagnostic criteria can aid clinicians in the diagnosis of this erratic condition.
Notes
Conflict of Interest
The author has no conflicts to disclose.