INTRODUCTION
In according to revision the Atlanta classification of acute pancreatitis (AP), AP classified 3 degrees of severity such as mild, moderately severe and severe AP. The definition of severity is based on the presence or absence of persistent organ failure which uses the modified Marshall scoring system and local and systemic complications. Mild AP defines as lack of organ failure and local/systemic complications. The organ failure that resolves within 48 hours and/or local or systemic complications defined as moderately severe AP. Severe AP is defined as when persistent single or multiple organ failure more than 48 hours [1]. Most patients with AP present with mild form and runs benign course that an uneventful recovery without organ dysfunctions and extremely low mortality. However, about 10~20% of patients are progress to severe AP [2]. It is important to determine severity early in the course of AP, because morbidity and mortality are quite different according to severity of the disease. Early identification of patients at risk for severe AP would appropriate critical care to reduce mortality. The mortality of severe AP is estimated about 10~20% despite of improvements in clinical care modalities [3,4]. In spite of many advances in diagnosis and management of AP, its pathogenesis is scarce although several mechanisms are proposed nowadays. It is known that the inappropriate activation of an inflammatory cascade is to be associated with development of severe AP [5,6]. However, the pathogenesis of severe AP is complicated. In this paper will be summarizes the proposed intra and extra acinar cell pathophysiologic mechanisms of severe AP focusing on development and progression.
PATHOGENESIS OF ACUTE PANCREATITIS
Several pathophysiologic mechanisms were proposed in AP but, the exact pathophysiologic mechanism is not well demonstrated. After the initial injury to the pancreatic acinar cell, whatever the triggering factor, events take a similar path for all patients with AP. Intra-acinar cellular trypsinogen activate to trypsin which then activates the other inactive proteases like domino and subsequent autodigestion have been regarded as the key pathogenesis of AP from Chiari who proposed the autodigestion theory in 1896 [7]. However, the AP is regarded as a kind of an inflammatory disorder induced by many various causes nowadays. The intra cellular trypsinogen activation only part of the pathogenesis of AP. Inflammatory mediators is another very important pathophysiologic mechanism of AP [8]. In addition, Dawra et al. [9] reported that intra-acinar trypsinogen activation contributes to early acinar injury, but local and systemic inflammation progresses independently during pancreatitis. The intra cellular inflammatory response which occurred as independently of trypsinogen activation may be a crucial pathophysiologic mechanism of pancreatitis [9]. The inflammatory response and mediators are playing an important role in the development and progression of AP. The progression of AP can be viewed as a three-phase continuum: local inflammation of the pancreas, a generalized inflammatory response, and the final stage of sepsis, with multiple organ damage [10]. Initial phase is characterized by premature activation of zymogen granules releasing active enzymes which cause acinar cell injury, autodigestion of pancreas and peripancreatic tissue. In second phase, recruited neutrophils in the pancreas induce intrapancreatic and local inflammatory reaction mediated by cytokines which are released from neutrophils. In third phase, activated proteolytic enzymes and many inflammatory cytokines lead to systemic inflammatory response syndrome (SIRS), acute respiratory distress syndrome (ARDS) and multiorgan failure (MOF). The disease process can extend to any of the three phases, and is often resolved after the local inflammatory process, resulting in mild AP.
THE FACTORS FOR INFLUENCE OF SEVERITY OF AP
1. The role of leukocyte
Leukocytes play an important role in the progression of the disease from a local inflammatory to SIRS early event during severe AP [11-13]. It is known that leukocytes are hallmark of inflammation emigrate to pancreas under the influence of chemotactic agents in early stage of AP and the degree of leukocyte infiltration was related to progress of severe AP [14]. The excessive leukocyte stimulation involves in the pathogenesis of severe AP and leukocyte proposed as a prognostic marker in AP [14]. Resulting from the inappropriate intra cellular activation of trypsinogen, autodigestion of pancreas tissue, resulting in necrosis of acini, peripancreatic fat necrosis and necrotizing vasculitis. The result of autodigestion induces excess recruitment of leukocytes to the pancreas. The leukocytes play an important role in release of pro-inflammatory cytokines and oxygen derived free radicals influence to development of acinar necrosis. These mediators are known as important elements which lead local inflammation to systemic inflammatory response and eventually MOF in AP [15]. It was known that the leukocyte depletion or impaired leukocyte activity shows that ameliorate the severity of AP in experimental model. Therefore, the leukocyte is one of an important role and influence factor for the disease pathogenesis, progression and eventually severity [16]. The adhesion molecules, inter-cellular adhesion molecule 1 (ICAM-1), vascular adhesive molecule 1 (VCAM-1), and selectine which require to neutrophil migration and adhesion in inflammatory area are upregulated in AP. They are also key role of AP progression from local to systemic disease as well as organ failure [17-19]. The ICAM-1 leads to leukocyte adhesion, increased capillary permeability, and reduced capillary blood flow velocity, and causes pancreatic microcirculatory disturbance through leukocyte-endothelial cell interaction [20,21]. The degree of microcirculation derangement is related with concentration of adhesion molecules and the peripheral blood neutrophil ICAM-1 expression is significant increase at early stage in severe AP but not in mild AP [17,22]. In addition, the inhibition of ICAM-1 or VCAM-1 by monoclonal antibody and use of antineutrophil antibody can attenuates the severity of AP and lung injury [23-25]. The early measurement of serum ICAM-1 levels within 24 hours can distinguish severe AP from mild AP. Therefore, the ICAM-1 may be a simple, rapid, reliable and early predictable marker for severe AP [26].
2. The role of cytokines and chemokines
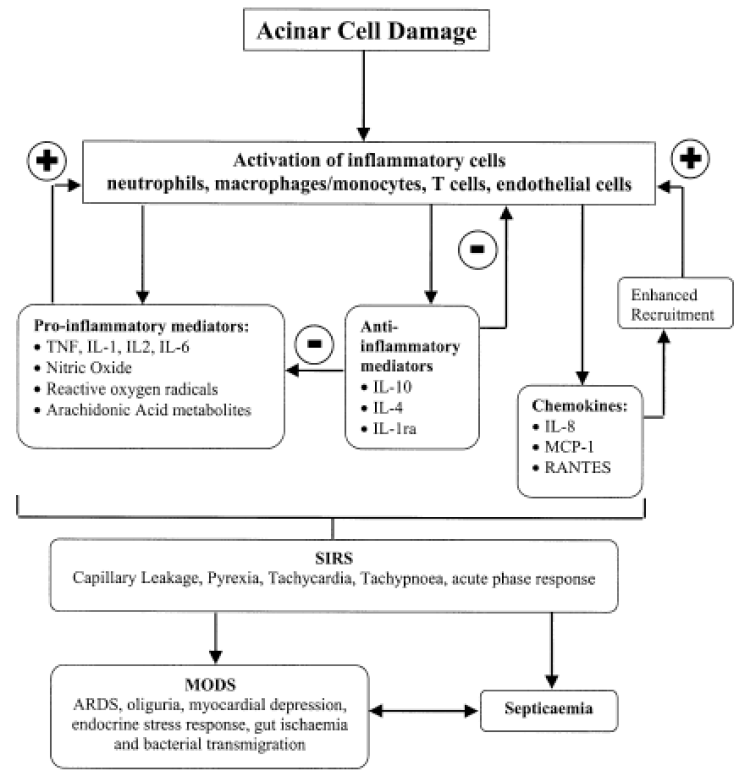
Cytokines are low molecular weight glycoproteins that are secreted by antigen presenting cells and act as mediators of immunity and inflammation [27]. In 1988, Rinderknecht was first hypothesized that cytokines may play an important pathophysiologic role in AP and suggested that inappropriate activation of the immune system might increase the disease severity [14]. It was reported that acinar cell itself like leukocyte can produce cytokine such as tumor necrosis factor α (TNF-α) in AP and this cytokine influence the patterns of acinar cell death [28]. Activated neutrophils in AP could induce massive release of cytokines and reactive oxygen species generation. The cytokines that released from leukocyte induce neutrophils accumulation in vital organs. Vital organs were damaged cause by neutrophils and the neutrophils in the damaged vital organ can reinduction of cytokines. The reinducted cytokines are contributed to induce neutrophils accumulate in vital organs by second attack (vicious cycle) eventually MOF in AP [15]. It is known that trypsin act as a potent mediator of the cytokines and also another inappropriate activated proteases release into bloodstream stimulate the production of cytokines which can leads to SIRS in AP [29,30]. The cytokine levels in blood were closely associated with the severity of illness on admission, the magnitude of MOF as well as with outcome. In human study, it was known that sustained release of both pro- and anti-inflammatory cytokines associate with severe acute pancreatitis and early MOF [31]. TNF-α that mainly produced in macrophages and monocytes is interact with other cytokines and has important role of inflammation and shock. The TNF-α is released from pancreas acinar cell in early course of AP and the level of TNF-α in blood have increased in patients in AP with MOF [28,32]. The proposed mechanisms of TNF-α induced pancreas injury are direct pancreas duct cell injury, formation of microthrombus, ischemia, hemorrhage, necrosis and edema [33]. The neutralization of TNF-α with an antibody produced a mild improvement in the parameters and can significant reduction in mortality and enhance the therapeutic effectiveness of octreotide for treatment of necrotizing AP in experimental studies [28,34,35]. Interleukin (IL)-1 which mainly produced from macrophage is important mediator of AP. In usually, TNF-α and IL-1 are regarded as first line cytokines. Therefore, IL-1 is important role in the early phase of AP and its level correlated with the severity and IL-1 levels increased in AP, especially in case of organ failure [6]. IL-1β which formed from IL-1 mediation of IL-1 convertase interacts with TNF-α to induce or aggravate organ injury. IL-1β is strongly associated with the development of SIRS. In addition, IL-1 antagonist and IL-1β activating enzyme inhibitors are decrease mortality and histologic grading in experimental severe AP [36]. IL-6 which produced by various cells but mainly generated from monocyte stimulated by TNF-α and IL-1β, its levels are increased in AP especially, in case of complicated AP [12,37]. Therefore, the IL-6 levels are correlated with disease severity and negatively correlated with prognosis. IL-6 can use an excellent predictor of disease severity in early phase of AP, because the IL-6 stimulates the synthesis of acute phase proteins, including C reactive protein, from hepatocytes [38]. IL-10 is an anti-inflammatory cytokine that inhibit the release of pro-inflammatory cytokines such as, IL-1β, TNF-α, and IL-6. Pretreated IL-10 agonist reduced the inflammatory response and mortality in experimental AP model [39]. The serum levels of IL-10 on the first day of the AP are higher in mild cases than in severe AP [40]. Therefore, the IL-10 levels are reverse correlated with severity of AP and the IL-10 act as a protective role in AP. The IL-4 and IL-11 have also anti-inflammatory role and attenuate the histologic severity in the early course of AP [41]. Platelet activating factor (PAF) is a potent pro-inflammatory cytokines which regarded as an important mediator of the SIRS [42]. The roles of PAF are promotion of platelet adhesion, aggregation, formation of thrombus, increase in capillary permeability and increase of blood viscosity. Therefore the PAF is closely related with microcirculatory disturbance [43]. The powerful PAF antagonist, Lexipafant, showed a decrease the incidence of organ failure in experimental study. However, the phase III trial did not show any improvement in organ failure rate or mortality [44]. Chemokines are 8-10 kDa small cytokines which chemotactic effects of leukocyte into area of inflammation and infection. IL-8 is a potent neutrophic chemoattractant and an important role in ARDS. The levels of IL-8 are correlated with severity of AP [45,46]. Anti-human IL-8 antibody reduce lung injury in an experimental AP [47]. In summary, cytokines and chemokines are important role in initiation and progression of AP (Fig. 1) [48]. The role of Transcriptional factors.
3. The role of transcriptional factors
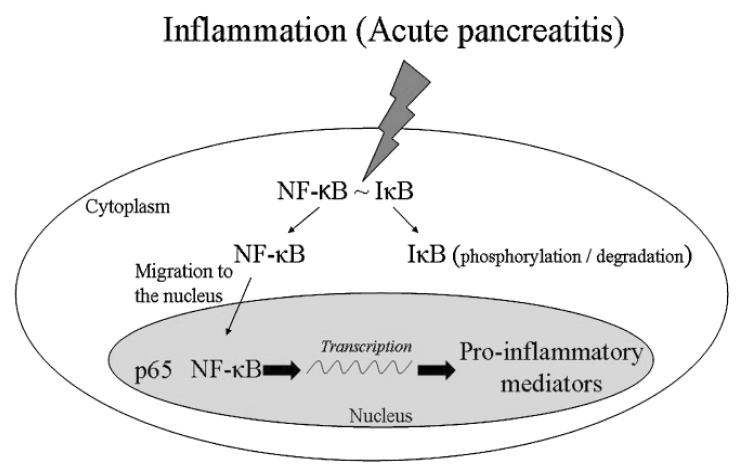
Nuclear factor-kappaB (NF-κB) that modulates the expression of most cytokines plays an important role in the initiation and progression of AP [49,50]. In normal condition, NF-κB is inactive state by inhibitors of κB (IκB). When cells are stimulated, IκB was degraded and then NF-κB is translocation into the nucleus and bind to its cognate DNA and increase the transcription of several proinflammatory genes (Fig. 2) [51]. NF-κB is activated in pancreas early phase of AP and subsequently distant tissues [52]. Intra acinar activation of NF-κB led to AP with local inflammation and systemic inflammatory response and the activation of NF-κB in vital organs is related with development of MOF in AP [53]. The activation of NF-κB in AP is independently of trypsinogen activation, interestingly. Dawra et al. [9] reported that trypsinogen is involved in pathologic activation and this leads to acinar cell death during early pancreatitis. However, progression of inflammation in AP is independent of trypsinogen activation. NF-κB activation is a key early event independent of trypsinogen activation and may be responsible for progression of local and systemic inflammation. This result arouses a doubt to traditional believed trypsin-centered pathophysiologic mechanism of AP. In addition, activation of inflammatory signaling mechanisms in acinar cell is crucial to pathogenesis of AP [9]. The NF-κB could upregulate the expression of cytokine including TNF-α, IL-1β, and chemokines in AP. It is known that blocking the NF-κB activation can reduce the pancreas and lung injury in experimental AP. In addition, inhibition of NF-κB has been shown to results attenuated pancreatitis response with severity inversely proportional to the degree of NF-κB inhibition [30,54,55].
THE ROLE OF APOPTOSIS, NECROSIS AND AUTOPHAGY
In generally, cell death can be classified according to morphological appearance, enzymological criteria, functional aspects and immunological characteristics. Morphologically, cell death can be classified as apoptosis, autophagy and necrosis. Apoptosis and autophagy are genetically controlled programmed cell death [56]. Apoptosis is physiological and programmed cell death which little or no induces inflammatory response. The mechanisms and pathway of apoptosis are relatively well described. The NF-κB, neutrophils and cytokines are affect the apoptosis in AP [28,49,57]. Apoptosis may be converted to necrosis by the recruitment of neutrophils due to the release of cytokine. The patterns of acinar cell death in AP are closed related with the disease severity. Necrotic cell death is correlated with severe AP whereas apoptotic cell death is associated with mild AP in experimental models. It is widely accepted that the severity of AP is inversely related to the extent of acinar cell apoptosis [58,59]. In contrast of apoptosis, necrosis can induces inflammatory response through intracellular contents are spillage to the extracellular space. In necrosis, released intracellular content are induces the immune response. When cells die from necrosis, damage associated molecular pattern (DAMP) molecules enter the circulation and activate innate immune cells. It is known that the inflammasome which DAMP receptors and an intracytosolic complex are initiated forms of inflammation [60,61]. The inflammasomes are required for the development of inflammation in acute pancreatitis and use of their antagonist demonstrated a reduced pancreatitis response [62]. Autophagy is the process by which cells recycle their own essential, redundant, or damaged organelles and macromolecular components. Autophagy also an adaptive response to sublethal stress, such as nutrient deprivation, that supplies the cell with metabolites [60]. Although the autophagic and lysosomal dysfunction is regarded as a kind of important initiating pathologic mechanism of pancreatitis, the role of autophagy to severity is not investigated now [63].
PATHOGENESIS OF ORGAN FAILURE IN ACUTE PANCREATITIS
Organ or multiorgan failure is major cause of death in the early phase, within 14 days, of AP. The failure rates of liver, heart, lung and kidney in AP are 48.9-60.7%, 15-50% and 14-35.8% respectively [64-67]. The pathogenesis of organ failure in AP is complicated. Proteases, inflammatory cells and its mediators which secreted from pancreas into blood circulation and impairment of microcirculations are explaining as the main contributed factors for organ failure in AP. The vasodilatation that mediated by mainly nitric oxide (NO) protective effect on circulatory derangement of AP. However, vasodilatation effect in AP associates with exacerbations AP cause of induction significant hypotension. The increase of the NO production and the decrease responsiveness to the endogenous vasoconstrictor are proposed mechanisms of hypotension in AP [68]. Acute lung injury (ALI) and ARDS are two main causes of death in AP related deaths. The pathogenesis of ALI in AP is also complicated. The characteristic features of ALI are increase of pulmonary microvascular permeability, with protein-rich exudate leaking into the alveolar space [69]. The proteases such as phospholipase A2 and elastase which released into systemic circulation in AP can induce the surfactant degradation and increased lung permeability. In addition, elastases which release into systemic circulation during AP can activate the inflammatory cells, NF-κB and TNF gene expression with subsequent pulmonary neutrophil recruitment and induce microvascular leakage. Upregulated cytokines, chemokines, adhesion molecules and pulmonary infiltration of neutrophils during of AP are important role of ALI progression in AP [70,71]. Acute kidney injury (AKI) is characterized by rapid loss of renal excretory function. The mortality rate has been markedly increased in case who have AKI in AP. Impairment of renal microcirculation, decrease renal perfusion pressure and hypovolemia play a role in the AP with AKI. Renal proximal tubule cells could metabolize the phospholipase A2 in AP. Pancreatic phospholipase A2 was rapidly deposited in renal tubular cells in AP and can induce renal tubular cell damage [72]. Reduce the blood supply to the kidneys in AP due to increase intra-abdominal pressure leading to AKI through low perfusion and ischemia. High level of blood trypsin in AP can release vasoactive polypeptide that induce renal toxicity and also cause a systemic hypercoagulabable state which reduce renal function by coagulation, thrombus formation in the renal blood vessels [73-75].
PATHOGENESIS OF PANCREATIC INFECTION
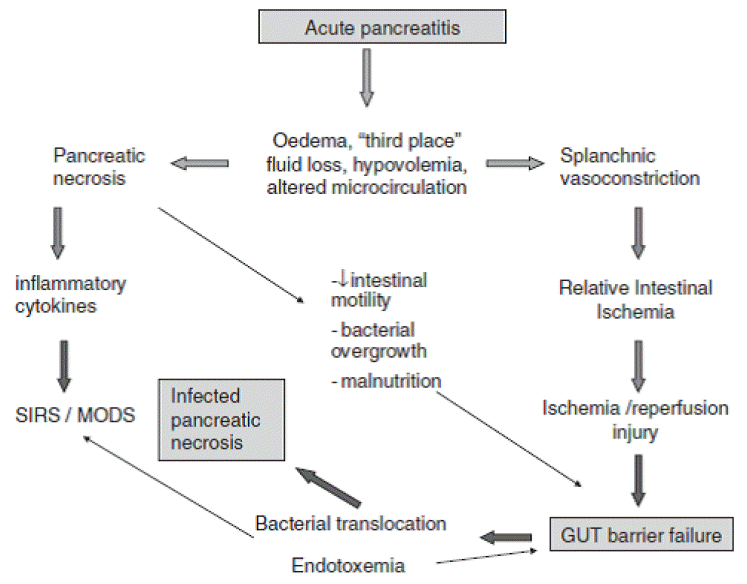
Infection, including infected pancreatic necrosis and fugal infection, is most common cause of late phase (2 weeks later the onset of disease) death in AP. The main pathways of infection into pancreas are hematogenous, lymphatics and transmural through the colonic wall, via ascites, biliary duct and pancreatic duct. The colon seems to be the main source of infection in AP, considering that most pathogens in pancreatic infection are gram-negative bacteria of gastrointestinal origin. In usually bacterial translocation from intestine is enhanced in AP and it leading to transperitoneal infection in AP. Bacterial overgrowth, delayed gut motility, impairment of the gut immune system and damaged mucosal barrier are involving factors of promotes bacterial translocation [76]. The gut plays a main role in infection in AP. The gut barrier dysfunction which is characterized by damage to the gut epithelium and intestinal tight junctions, resulting in increased intestinal permeability present in 60% of patients with AP [77]. The gut barrier dysfunction facilitate as well as the bacterial translocation and endotoxin into the extra-intestinal site. The proposed pathogenesis of gut barrier dysfunction were mucosal ischemia, disruption of mucosal epithelial integrity, reperfusion injury, disruption of intestinal bacterial ecology, hypovolemia, impaired mucosal immunity, endotoxemia and cytokines. In addition, loss of the unstirred mucus layer but not villous injury is associated with gut barrier dysfunction in AP (Fig. 3) [78-80]. Enteral nutrition is associated with reduced mortality, low septic complications and hospital stay in severe AP. These beneficial effects of enteral nutrition in AP are associated with protection of gut mucosal barrier to prevent bacterial translocation form intestine [81]. The prevalence of gut barrier dysfunction is affected by patients age not by the disease severity, interestingly. In meta-analysis, there is a 2% decrease in the prevalence of gut barrier dysfunction for every 1-year increase in patient age [77]. Gut barrier dysfunction is closely related to infectious complications in AP. Therefore, therapeutic strategies which to maintain the gut barrier in severe AP are important to reduce the infectious complication and mortality.
CONCLUSIONS
The majority of patients with AP runs mild course, but some of them progress to a severe course. Although the knowledge of intra and extra acinar cellular mechanisms of AP has been evolving over the decades, the mechanisms which why the AP develops to become a severe form in some patients is not made sufficiently clear. The pathophysiology of severe AP is complicated and caused by combines of manifold factors. During the initiation and progression of AP, inflammatory cells, cytokines, chemokines, transcription factors, gut barrier dysfunction and patterns of acinar cell death interact and mediate the development of severe AP. It is important to understanding the pathophysiological mechanisms of severe AP to reduce morbidity and mortality through the early detection the patients who are at risk for developing severe AP. Therefore, the further studies will be focus to searching salient culprit or mechanism which can involve in severe AP.