서 론
담도암(bile duct cancer, BDC)은 간외담관에 발생하는 가장 흔한 악성질환으로, 그 예후가 매우 불량하다, 그것은 증상이 늦게 발현하여 절제율이 낮기 때문이다. 또한 보존적 항암치료나 방사선 치료의 치료 성적이 만족스럽지 않다[1,2].
Ursodeoxycholic acid (UDCA)는 친수성 담즙산으로 원발성 담즙성 경변증의 치료제로 사용되어 왔다. 혐수성 담즙산과는 달리, UDCA는 세포자멸사(apotosis)를 억제하여 담관세포 및 간세포를 보호한다. 이는 UDCA가 epidermal growth factor receptor (EGFR)/mitogen-activated protein kinase (MAPK) 생존 경로를 활성화하여 미토콘드리아의 기능저하를 막고, pro-apoptotic cascade의 활성화를 억제하기 때문으로 추정된다[3,4]. 하지만 UDCA는 논란이 있지만 정상세포와는 달리 암세포에서는 세포자멸사를 유발하는 것으로 알려져 있다. 또한 UDCA가 BDC 발생의 위험을 낮추는가에 대해서는 여전히 논란의 여지가 있으나, 일부 역학관련 연구에서 그 가능성을 시사하고 있다[5,6]. 최근의 본 교실의 연구에서, UDCA가 BDC 세포에서 세포자멸사와 p53의 활성화를 유발하여 deoxycholic acid (DCA)가 유발한 EGFR-extracellular signal-regulated kinase (ERK) signaling, cyclooxygenase Inhibitor-2 (COX-2), phosphoinositide 3-kinases (PI3K) - protein kinase B (PKB, AKT) signaling을 억제하였고, 결국에 BDC 세포의 성장과 침윤을 억제함을 확인하였다[7].
상피-중간엽 전환(epithelial-mesenchymal transition, EMT)은 상피세포가 점차 중간엽세포의 구조적이고 기능적인 특징을 도입하는 가역적인 역동성 과정이다[8-10]. EMT는 종양세포의 원발부위에서 전이와 침윤의 초기과정이기도 하다. 주요한 EMT 과정은 상피 표현형(epithelial phenotype)을 억제하여 중간엽 표현형(mesenchymal phenotype)이 활성화되도록 유전자발현을 변환시키는 것이다[11,12]. BDC의 진행과 전이에 있어서, EMT의 특징의 존재와 병리적 심각도가 서로 관련이 깊음이 입증되었다. 즉, 상피 marker (E-cadherin 등)의 상실과 중간엽 marker (Slug, S100A4, vimentin, N-cadherin 등)의 취득이 BDC의 공격적인 특징과 관련이 컸다[13-16]. 최근 본 교실의 연구에서 UDCA가 BDC 세포에서 EMT의 억제를 통하여 세포자멸사 유도와 BDC 세포의 전이성을 억제함으로써 항종양 효과의 증거를 제시한 바 있다[17].
활성산소(reactive oxygen species, ROS)는 산소의 환원에 의해 발생하는데, 악성 세포의 세포주기 진행, 증식, 생존, 세포자멸사, 그리고 혈관생성에 영향을 준다[18]. Raf, Myc, 그리고 cyclin E와 같은 발암유전자의 과발현은 ROS 생성의 증가와 연관이 있으며[19], 또한 일부 연구에서 ROS는 EMT 과정의 중개자(mediator) 또는 조율자(modulator)로 여겨지고 있다[20-22]. 한편, UDCA는 다양한 암세포에서 ROS의 생성을 억제하는 것으로 알려져 있다[23-25]. 더욱이 BDC에서는 UDCA가 ROS와 밀접한 관련이 있는 PI3K/AKT 경로와 MAPK/ERK1/2 signaling을 억제하므로[7], 어떤 방식으로도 ROS 생성 및 관련 경로에 영향을 줄 가능성이 매우 높다. 그러나, UDCA가 BDC 세포에서 ROS 생성 및 제거에 어떠한 영향을 주는지에 대해서는 알려진 바가 없다.
따라서, UDCA가 BDC 발생, 성장 및 침윤에 관여하는 ROS 생성 및 ROS와 관련이 있는 단백질에 어떠한 영향을 미치는지 알아보고자 한다. 또한 ROS가 촉발한 EMT를 UDCA가 억제하는지 알아보고자 한다. 궁극적으로 BDC의 성장 및 침윤을 억제할 수 있는 항암제로서, 또는 보조치료제로서 기여할 수 있는 가능성을 확인하고자 본 연구를 시행하였다.
방 법
1. 재료
Roswell Park Memorial Institute (RPMI) 1640 medium, fetal bovine serum (FBS), penicillin-streptomycin, trypsin 및 sodium bicarbonate (Gibco, Grand Island, NY, USA)의 제품을 사용하였다. Dimethyl sul foxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), human EGF, 그리고 UDCA는 Sigma Chemicals (St. Louis, MO, USA)에서 구입하였다. Goat anti-rabbit IgG-horseradish peroxidase (HRP)는 Santa Cruz Biotechnology (Santa Cruz, CA, USA) 제품이었다. Western Blot Hyper HRP substrates는 Takara (Kusatsu, Shiga, Japan)에서, gefitinib은 Roche Diagnostics (Indianapolis, IN, USA)에서 구입하였다. Phospho-STAT3, STAT3, peroxiredoxin 2 (PRX2), NADPH oxidase 2 (NOX2), NADPH oxidase 4 (NOX4), catalase, SOD2, E-cadherin, N-cadherin, 그리고 β-actin 항체는 Cell Signaling Technology (Danvers, MA, USA) 제품을 사용하였다.
2. 세포배양
SNU-245 세포주는 분화도가 좋으며, 특정 유전자의 주요 돌연변이가 없는 총담관에서 추출한 동양인 BDC 세포로서, Korean Cell Line Bank (KCLB)에서 공급하였다. KCLB는 세균 및 미코플라스마 오염 없음과 short tandem repeat (STR)을 확인하고 보증한다. SNU-245 세포는 2 mM glutamine, 10% FBS, 1.5 g/L sodium bicarbonate, 100 μg/mL streptomycin, 그리고 100 IU/mL penicillin이 첨가된 RPMI 1640 배지를 사용하여 배양하였다. 배지는 주당 2회 교체되었으며, 37°C로 온도를 유지하고, 5% CO2가 공급되는 배양기에서 세포는 배양되었다. 세포가 배양접시에 충만되면 EDTA (1.0 g/L)와 trypsin (2.5 g/L)을 사용하여 분리하여 계대배양을 시행하였다.
3. MTT assays
MTT assays는 세포의 성장을 측정하기 위해 이전에 기술한 방법대로 진행하였다[17]. RPMI 배지에 세포들을 96-well 용기내에 5×104 cells/mL의 밀도로 분주하고 24시간 배양하였다. 이후 정해진 농도의 UDCA, DCA, 그리고 insulin-like growth factor 1 (IGF-1)을 처리 후, serum-free medium (SFM) 환경에서 24 또는 48 시간 배양하였다. 그 후 MTT (0.5 mg/mL)를 각 well에 처리하고 추가로 4시간 배양을 하였다. 이후 배지를 제거하고, 100 μL의 DMSO를 각 well에 첨가하였다. 발색반응(colorimetric response)은 ELX800 (Biotek, Winooski, VT, USA)을 사용하여 570 nm 파장으로 측정하였다.
4. ROS 측정
ROS는 celluar ROS assay kit (abcam, Waltham, MA, USA)를 사용하여 측정하였다. BDC 세포를 2×105 cells/mL 밀도로 6-well plat에 분주하여 200 μM UDCA, 200 μM DCA 또는 100 nM IGF-1으로 처리하여 24시간을 두었다. 세포들은 이후 포집되어 phosphate buffered saline (PBS)로 세척하였다. 96-well plate에 well당 25,000 세포들이 담기도록 분주하고 25 μM DCF-DA를 37°C에서 45분간 처리하였다. 형광강도(fluorescence intensity)는 fluorescence plate reader (Molecular Devices, San Jose, CA, USA)를 사용하여 485/535 (Ex/Em) nm의 파장으로 측정하였다.
5. Hydrogen peroxide/peroxidase assay
Hydrogen peroxide/peroxidase assay kit (Abcam)를 사용하여 제조사의 지시서에 따라 hydrogen peroxide와 peroxidase를 측정하였다. BDC 세포를 2×105 cells/mL 밀도로 6-well plate에 분주하고, 200 μM UDCA 또는 200 μM DCA를 처리하고 24시간 배양하였다. 그후 세포를 포집하여 PBS로 세척하였다. 이후 5분간 얼음위에서 세포들을 다시 assay buffer를 사용하여 풀고, 15,000 rpm으로 4°C에서 5분간 원심분리 하였다. 세포들을 reaction buffer와 섞은 후 상온에서 10분간 암실에 두었다. 발색반응은 ELX800 (Biotek)을 사용하여 570 nm 파장으로 측정하였다.
6. Cell transfection
BDC 세포를 2×105 cells/mL의 밀도로 6-well plate에 분주하고, SiRNA 또는 ShRNA oligonucleotide를 세포내 transfection하였다. 표적 STAT3, PRX2와 non-silencing control siRNA는 Invitrogen (Carlsbad, CA, USA)에서 제조되었다. 3 μL lipofectamine 2000 (Invitrogen)이 혼합된 500 μL serum-free OPTI MEM (Gibco, Grand Island, NY, USA)내에 용해된 20 pmol SiRNA oligomer가 각각의 transfection에 사용되었다. 세포들은 1.5 mL의 항생제가 포함되지 않은 성장배지에서 48시간 동안 transfection이 진행되었다. 이후 1% bovine serum albumin (BSA; Sigma Chemicals)이 포함된 SFM으로 24시간 배양되었다. NOX4와 non-targeting control ShRNA는 GenePharm (Shanghai, China) 제품을 사용하였다. 이후 transfection은 제조사의 지시서에 따라서 시행되었다. 각 transfection의 영향은 western blot으로 측정하였다.
7. Western blot assays
Western blot assay는 이전에 기술한 방식대로 진행되었다[17]. SNU-245세포의 중식이 세포배양접시 내에서 90% 수준에 이르면 분주하여 지정된 농도의 UDCA, DCA 또는 IGF-1를 24 또는 48시간 처치하였다. 세포를 포집한 후 PBS로 세척하였다. 세포들은 RIPA buffer (Sigma Chemicals)로 용해시킨 후 20분간 15,000 rpm으로 원심 분리하였다. Bradford assays (Sigma Chemicals)로 추출된 단백질의 양을 측정하였다. Blot은 상온에서 blocking solution으로 처리하고 phosphorylated signal transducer and activator of transcription 3 (pSTAT3) (1:1,000 dilution), STAT3 (1:1,000 dilution), PRX2 (1:1,000 dilution), NOX2 (1:1,000 dilution), NOX4 (1:1,000 dilution), catalase (1:1,000 dilution), SOD2 (1:1,000 dilution), E-cadherin (1:1,000 dilution), N-cadherin (1:1,000 dilution), 또는 β-actin (1:1,000 dilution)에 대한 rabbit polyclonal antibody가 포함된 5% BSA solution에 반응하도록 밤새 4°C로 둔다. Nitrocellulose membrane은 30분간 상온에서 TBS-T로 세척 후 goat anti-rabbit IgG-HRP (1:8,000 dilution)로 처리한다. Western Blot Hyper HRP substrate가 특정 band를 검출하기 위해 사용되었다. 각 band의 시그널 강도는 ImageJ (Version 1.43; National Institute of Health, Bethesda, MD, USA)로 측정하였다.
8. 통계분석
모든 실험은 정해진 같은 실험조건에서 다른 sample을 사용하여 최소 3회 이상 실행되었다. 모든 기술된 데이터는 평균±표준편차(mean±standard deviation)로 표시되었다. 데이터는 normality test (Skewness and Kurtosis statistics) 후에 parametric distribution을 확인하였다. 두 개의 unpaired group은 Student’s t-test, 세 개 이상의 group은 one-way ANOVA 후 Tukey post hoc test를 시행하여 비교하였다. p-value <0.05를 통계적으로 의미 있다고 판정하였다. IBM-SPSS version 27 (IBM Corp., Armonk, NY, USA)을 사용하여 통계 분석을 하였다.
결 과
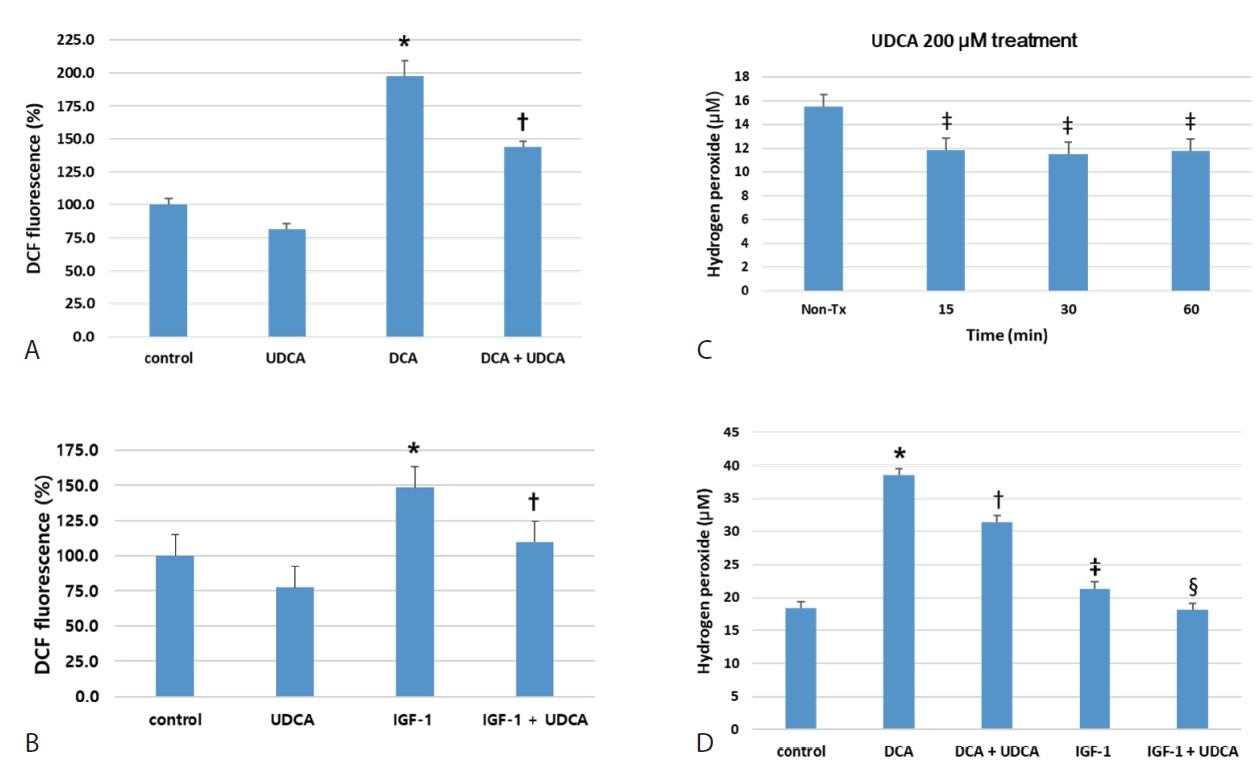
1. UDCA는 BDC 세포에서 ROS의 생성을 억제한다.
UDCA가 BDC 세포에서 DCA 및 IGF-1이 유발한 ROS의 생성을 억제하는지 ROS assay kit을 사용하여 확인하였다. UDCA와 DCA는 24시간 처리되었고, IGF-1은 UDCA 24시간 선처리 후 마지막 15분간 처치하였다. 200 μM DCA를 처리한 SNU-245 세포의 ROS의 생성은 2배 수준으로 상승하였으나, 200 μM UDCA와 함께 DCA를 처치하였을 때는 의미 있게 ROS의 생성이 낮아지는 것을 확인하였다(Fig. 1A). 100 nM IGF-1으로 처리 후, SNU-245 세포의 증가된 ROS의 생성 또한 UDCA로 사전 처치 시 의미 있게 낮아지는 것을 발견 하였다(Fig. 1B). 대표적인 ROS인 H2O2 생성을 측정할 수 있는 hydrogen peroxide assay에서도 200 μm UDCA는 BDC 세포의 H2O2의 생성을 단시간내 의미 있게 억제하였을 뿐 아니라(Fig. 1C), ROS assay와 양상과 마찬가지로 200 μM DCA와 100 nM IGF-1 유발한 H2O2 생성을 24시간 후 의미 있게 억제 하였다(Fig. 1D).
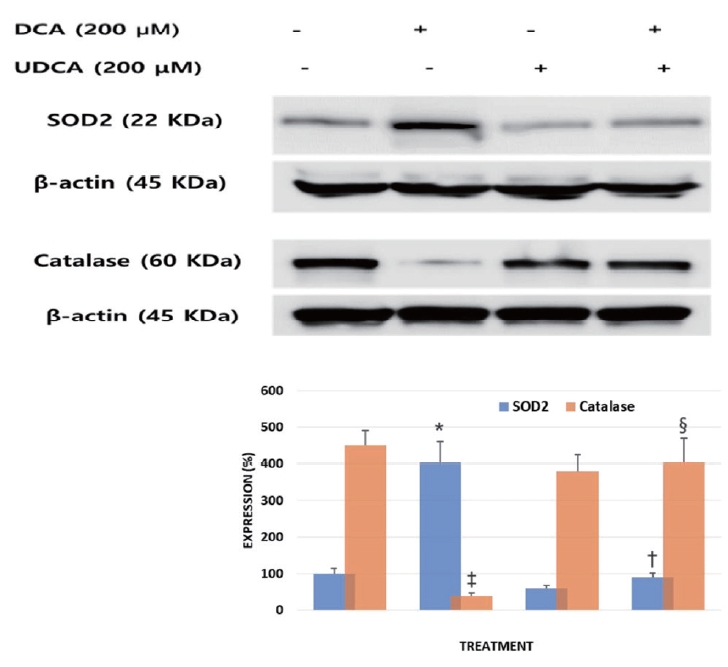
2. BDC 세포에서 UDCA는 MnSOD (superoxide dismutase 2, SOD2)를 억제하고, catalase를 활성화 한다.
UDCA가 BDC 세포에서 대표적인 ROS중 H2O2의 생성에 관여하는 MnSOD (SOD2)와 제거에 관여하는 catalase에 어떠한 영향을 미치는지 알아보았다. SNU-245 세포는 protocol에 따라 200 μM DCA 또는 200 μM UDCA를 처리 후 SFM에서 24시간 배양되었으며, 이후 방법에서 기술한 것처럼 western blotting을 시행하였다. DCA 처치에 의해 증가된 SOD2의 발현은 UDCA 처치에 의해 억제됨을 확인하였고, DCA 자극에 의해 억제되었던 catalase의 발현은 UDCA 처치에 의해 회복됨을 발견하였다(Fig. 2). 즉, UDCA는 BDC 세포에서 직접 H2O2의 생성을 억제하고 생성된 H2O2 제거에 관여함을 알 수 있었다.
3. UDCA는 BDC 세포에서 PRX2 및 STAT3의 발현을 억제한다.
이 실험은 BDC 세포에서 ROS 생성이 증가함에 따른 EMT 증가에 관여하는 PRX2 및 STAT3의 발현에 UDCA가 어떠한 영향을 주는지 알아보기 위해 western blot assay를 사용하여 시행되었다. 세포는 200 μM UDCA 또는 200 μM DCA를 처리 후 serum free media (SFM)에서 24 또는 48시간 배양되었다. DCA를 24시간 처리 후에 PRX2의 발현은 의미 있게 상승하였으며, UDCA는 증가된 PRX2의 발현을 매우 의미 있게 억제하였다. pSTAT3 또한 비슷한 양상으로 UDCA에 의하여 의미 있게 억제되었다(Fig. 3).
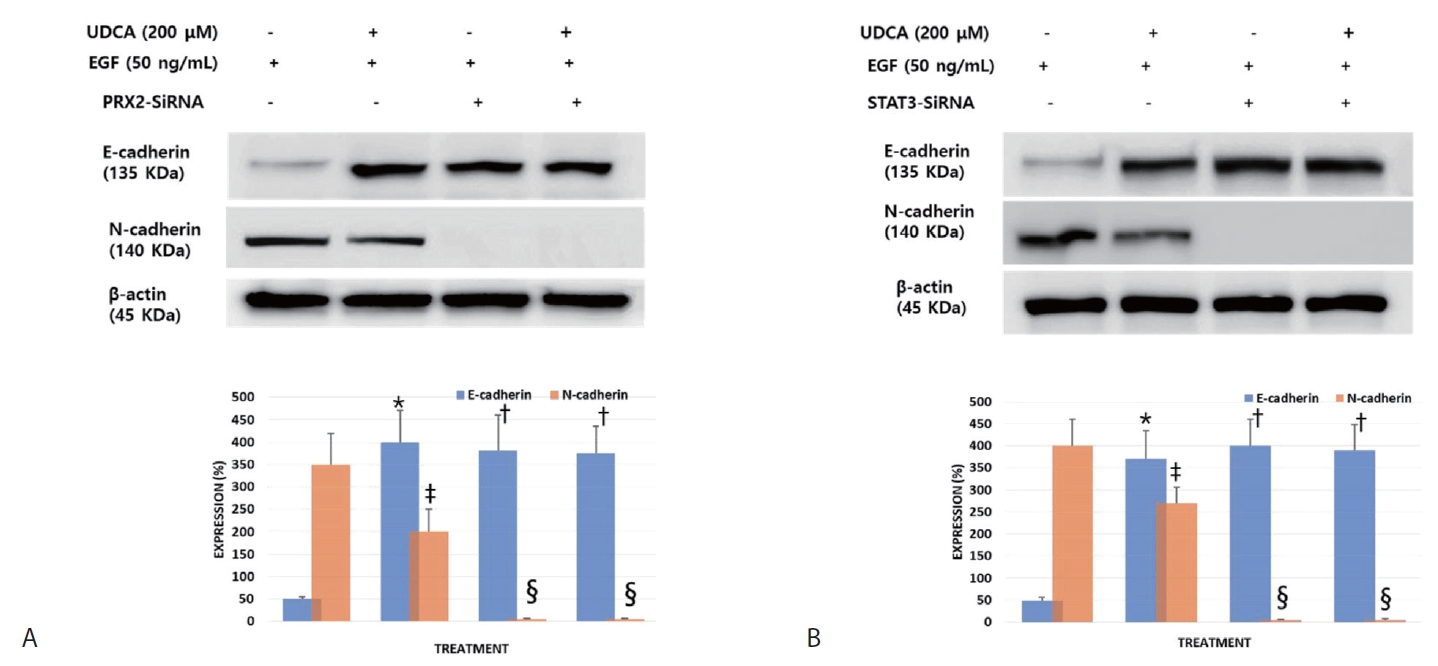
4. BDC 세포에서 UDCA가 EMT를 억제하는데 있어서 PRX2 및 STAT3가 관여한다.
이 실험은 BDC 세포에서 UDCA는 EGF가 억제한 E-cadherin을 복구하고, 증가시킨 N-cadherin을 억제하는지 확인하고, PRX2 및 STAT3가 이 과정에 얼마나 관여하는지 확인하기 위해 western blot assay 및 siRNA silencing을 이용하여 시행되었다. 세포는 200 μM UDCA 또는 50 ng/mL EGF 처리 후 SFM에서 48시간 배양되었다. EGF의 자극에 의해 증가된 N-cadherin의 발현은 UDCA 처치에 의해 억제되었으며, PRX2와 STA3을 silence 시킨 조건에서는 UDCA 처치와 상관없이 EGF 자극에 의해 증가한 N-cadherin의 발현은 전혀 나타나지 않았다. E-cadherin은 EGF의 자극에 의해 억제된 상태에서 UDCA 처치 후 다시 발현이 증가함을 볼 수 있지만, PRX2와 STAT3을 silence 시킨 조건에서는 UDCA 처치와 상관없이 EGF 자극으로 감소하였던 E-cadherin의 발현이 UDCA 처치한 수준으로 회복됨을 볼 수 있었다(Fig. 4).
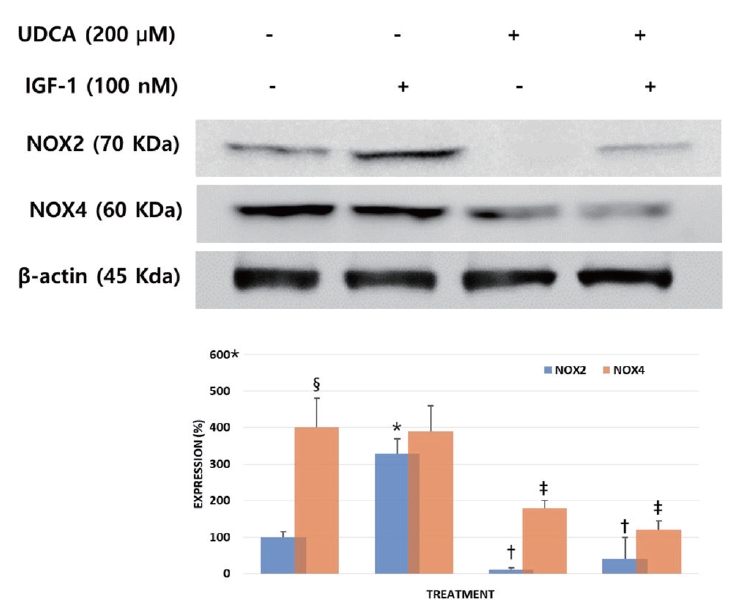
5. BDC 세포에서 UDCA는 NOX2와 NOX4의 발현을 억제한다.
이 실험은 BDC 세포에서 IGF-1에 의해 활성화된 NOX2와 NOX4에 UDCA가 어떠한 영향을 주는지 확인하기 위해 western blot assay를 사용하여 시행되었다, 세포는 protocol에 따라 IGF-1은 UDCA 24시간 선처리 후, 필요시 마지막 15분간 처치하였다. IGF-1 처리 후 NOX2의 발현은 의미 있게 증가하였으며, UDCA 처리 후 IGF-1의 처치가 없는 조건에서 발현이 억제가 되었다. UDCA와 IGF-1을 함께 처치한 경우도 IGF-1이 유발한 NOX2 발현의 증가를 비처치 대조군(non-treatment control) 이하로 의미 있게 낮추었다. 비처치 BDC 세포에서 NOX4의 발현은 NOX2에 비해 현저히 강하게 발현되었으며, IGF-1 처치시 NOX-2에 비해 발현의 증가가 관찰되지 않았다. 그러나 24시간 UDCA 처리 후 IGF-1의 사전 처치가 없는 조건에서 발현을 의미 있게 낮추었고, IGF-1의 사전 처치 후 24시간 UDCA 처리한 경우도 비처치 대조군 이하로 억제하였다(Fig. 5).
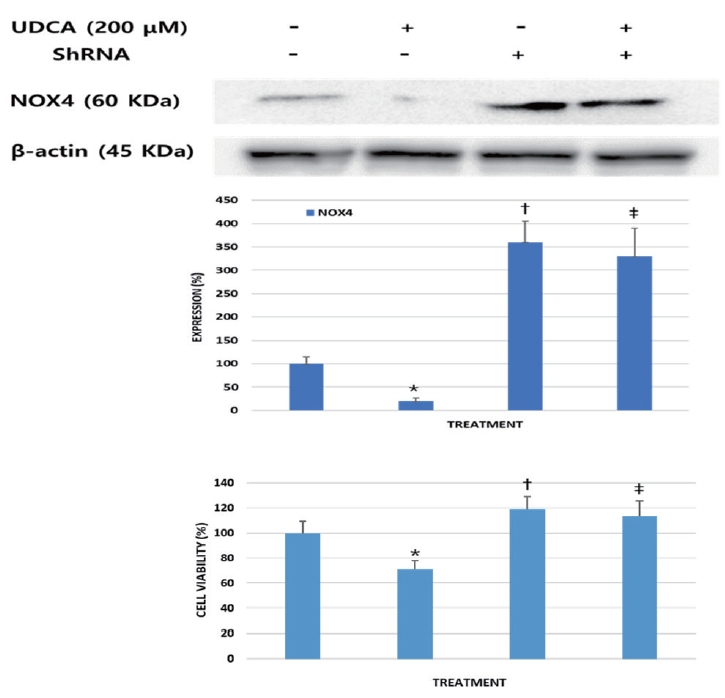
6. BDC 세포에서 NOX4의 과발현은 UDCA의 항종양 효과를 약화시킨다.
NOX4-ShRNA를 사용하여 NOX-4의 과발현을 유도하여, UDCA의 항암효과에 어떠한 영향을 주는지 확인하기 위해 western blotting과 MTT assay를 시행하였다. SNU-245 세포를 200 μM UDCA로 처치 후 NOX-4의 발현이 감소하고, 세포 생존력(cell viability)이 의미 있게 감소하였다. NOX4 과발현을 위해 shRNA가 transfection된 세포는 비처치군에 비해 세포 생존력이 의미 있게 증가되었고, UDCA에 의한 세포 생존력 억제효과도 약화되었다(Fig. 6).
고 찰
UDCA는 여러 암세포에서 세포자멸사를 유도하여 세포증식을 억제하는 효과가 있으며, 이는 많은 세포실험과 이종이식 동물 모델(xenograft model)을 이용한 연구에서 증명되었다[26,27]. 최근 본 연구자들은 BDC 세포에서 UDCA가 p53의 활성화하며, EGFR-ERK pathway, COX-2 및 PI3K-AKT pathway의 활성화의 억제를 통해 세포자멸사를 유도하여, 증식을 억제하고 공격성을 약화시키는 것을 확인하였다[7]. 본 연구자의 또다른 후속 연구에서, UDCA는 E-cadherin의 활성화와 N-cadherin의 억제를 통해 BDC 세포의 EMT를 방해하고 공격성을 약화시킴을 증명하였다[17].
ROS는 산소분자의 환원에 따라 형성되는 radical과 ion 등으로 구성된다. ROS로 인한 세포내 핵산, 단백 및 지질의 손상은 악성화와 연관이 있다. 특히 ROS는 암세포의 주기, 증식, 생존, 세포자멸사, 혈관생성 등에 영향을 준다[28]. 여러 pathway에 작용하는 발암관련 유전자들은 ROS 생성의 증가에 따라 영향을 받는 것으로 알려져 있다[18,19]. 일부 연구에서 ROS는 EMT 과정의 중계자로 제시되고 있으나[20-22], EMT 촉진과 세포의 악성화에 있어서 ROS의 역할은 아직 규명되어야 할 부분이 많다. BDC 세포에서 UDCA는 PI3K-AKT pathway와 MAPK-ERK1/2 signaling을 억제하므로[7], ROS 생성 및 관련 pathway에 영향을 줄 가능성이 매우 높으나, UDCA가 BDC 세포에서 ROS 생성에 어떤 영향을 주는지 알려진 바가 없다. 더욱이 BDC 세포에서 UDCA에 의해 억제된 EMT가 ROS 생성의 억제에 따른 결과인지 알려진 바 없다. 따라서 이에 대한 의문을 해결하기 위해 본 연구가 진행되었다. 본 연구의 ROS assay 및 hydrogen peroxide assay에서, UDCA는 BDC 세포의 ROS 및 H2O2의 생성을 의미 있게 억제하였을 뿐 아니라 DCA와 IGF-1이 유발한 ROS와 hydrogen peroxide 생성을 억제함을 관찰할 수 있었다. 이러한 결과가 우연이 아님을 증명하기 위해 ROS 중 H2O2의 생성에 직접 관여하는 MnSOD (SOD2)와 제거에 관여하는 catalase에 UDCA가 어떠한 영향을 미치는지 확인한 결과, DCA 처치에 의해 증가된 SOD2의 발현은 UDCA 처치에 의해 억제되었고, DCA 자극에 의해 억제되었던 catalase의 발현은 UDCA 처치에 의해 회복됨을 확인하였다. 이 결과들을 종합하면, UDCA는 BDC 세포의 ROS 억제뿐 아니라 담관세포의 급만성 염증 및 악성 변화와 연관이 있는 DCA 및 IGF-1이 유발한 H2O2를 포함한 ROS 생성도 효과적으로 억제할 수 있음을 알 수 있었다.
PRX2는 H2O2와 같은 ROS에 노출되면 발현이 증가되어 ROS를 제거하는 항산화제의 역할을 하나[29,30], 지속적인 발현의 증가는 암줄기세포(cancer stem cell)의 항암제 내성과도 관련이 있다. 또한 H2O2의 노출에 이어 STAT3와 반응하여 phosphorylation과 oligomerization을 증대하며 STAT3의 전사기능(transcriptional function)을 강화한다[31,32]. STAT3는 정상세포에서는, cytokine 및 성장인자에 의한 signal에 반응하여, 세포질에 존재하던 STAT3는 핵 안으로 이동하여 세포의 발달, 분화, 생장, 생존, 신생혈관합성 그리고 면역기능 등을 위한 유전자들을 조절한다. 그러나 비정상적인 경우, STAT3는 암의 발생에 중요한 기능을 한다. 많은 악성종양에서 활성화된 STAT3가 발견되었으며 STAT3에 의해서 암을 유발하고 발전시키는 여러 다양한 유전자의 발현을 조절한다[33]. 특히 암줄기세포의 형성에 관여하는 SOX2를 활성화하며, 암세포의 침윤과 공격성을 좌우하는 EMT를 촉진하는 것으로 알려져 있다[34,35]. 본 연구에서 UDCA는 BDC 세포에서 증가된 PRX2의 발현을 의미 있게 억제하였을 뿐 아니라 인산화된 STAT3도 의미 있게 억제하였다. 이 결과는 UDCA가 BDC 세포에서 ROS 억제에 따라 연관된 PRX-2 및 STAT3 신호경로가 억제됨을 보여주는 증거이기도 하다.
UDCA는 BDC 세포의 EMT를 억제하는데, 이 과정에 PRX2 및 STAT3가 얼마나 관여하는지 확인하기 위해 SiRNA silencing assay를 활용하여 연구를 진행하였다. SiRNA를 사용하여 PRX2와 STAT3을 silencing 시키면, UDCA 처치와 상관없이 EGF 자극에 의해 증가한 N-cadherin의 발현은 전혀 나타나지 않았으며, E-cadherin 또한 PRX2와 STAT3을 silencing 조건에서는 UDCA 처치와 상관없이 EGF 자극에 의해 감소하였던 E-cadherin의 발현이 회복됨을 볼 수 있었다. 한편, 본 연구자들의 BDC 세포를 이용한 이전 연구에서, EGF는 용량 의존적으로 E-cadherin의 발현을 억제하고 N-cadherin의 발현을 증가함을 확인한 바 있다[17]. 이는 BDC 세포에서 PRX2와 STAT3가 EMT에 상당한 영향을 주는 인자임을 시사하는 결과이다. 즉, UDCA가 ROS를 억제하여 PRX2 및 STAT3의 활성화를 억누르고, 이는 EMT의 억제로 이어졌을 것을 가능성을 보여준다.
NOX는 superoxide (O2-) 생성 효소로 NADPH로부터 전자를 O2분자로 이동시키는 촉매작용을 한다[36]. NOX family member는 NOX1-5로 세분화되며 구분되는 특징을 지닌다. 많은 연구에서 NOX로 생성된 ROS는 MAPK, AKT, NF-κB 그리고 matrix metalloproteases 등의 종양 관련 biomarker의 활성화와 관련이 있다[37,38]. 특히, 증가된 NOX4의 유전자 및 단백 발현은 여러 암종에서 관찰되며. 치료의 타겟이 되고 있다[39,40]. 본 연구에서 UDCA는 BDC 세포에서 IGF-1에 의해 활성화된 증가된 NOX2와 NOX4의 발현을 모두 의미 있게 억제하였으며, 또한 이 결과는 UDCA가 BDC 세포에서 ROS를 유발하는 oxygen free radical의 생성을 NOX2와 NOX4 활성화의 방해를 통하여 직접 억제함을 보여준다. 또한 본 연구에서 BDC 세포의 NOX4의 발현이 NOX2에 비해 현저히 강하게 발현되었는데, NOX4가 BDC 세포에서 ROS 생성 및 공격성에 더 많은 역할을 할 가능성을 제시한다. 따라서 NOX4의 과발현이 UDCA의 항암효과에 어떠한 영향을 주는지 확인하기 위해 연구에서, UDCA로 처치한 BDC 세포의 NOX4의 발현이 감소하고, 세포 생존력이 의미 있게 감소함을 확인하였고, NOX4 과발현을 위해 ShRNA가 transfection된 세포는 비처치군에 비해 세포 생존력이 의미 있게 증가되었고, UDCA에 의한 세포 생존력 억제효과도 약화되었다. 이 결과는 BDC 세포에서 NOX4의 과발현이 암세포의 공격성에 주요한 인자임을 시사하며 향후 이를 치료의 목표로 삼아 연구가 필요함을 보여준다.
본 연구는 인간 BDC 세포주를 이용한 in vitro 연구로써, 생체에서의 실제 항암효과를 증명하는 데에는 한계가 있다. 따라서, BDC 이종이식 동물 모델을 이용하여 기존의 다른 항암제와 함께 또는 단독으로 UDCA의 ROS 및 EMT에 대한 영향과 실제 항암효과를 조사하는 연구를 향후 추가로 시행하고자 한다 . 또한 BDC에서 이미 입증된 기존 화학요법과의 UDCA의 시너지 효과를 평가하기 위한 다양한 임상 연구가 시행되기를 기대한다. 한편, 본 연구는 고분화도 BDC 세포주인 SNU-245 cells를 대상으로 진행된 바, 다양한 분화도와 돌연변이가 있는 다양한 종류의 BDC 세포주를 대상으로 연구가 필요할 것이며, 본 연구의 또 다른 한계점이 될 수 있다. 다만, 본 연구자들은 돌연변이에서 유래된 많은 변수를 피하기 위해 E-cadherin을 발현하고 p53, p15, p16, hMLH1, K-Ras의 돌연변이가 없는 wild BDC 세포를 평가하고자 하였다. 향후 연구에서는 다른 BDC 세포주를 대상으로 연구가 이루어지길 바란다.
결론적으로 UDCA는 BDC 발생, 성장 및 침윤에 관여하는 ROS의 생성을 억제하며, ROS 생성 및 제거에 관여하는 여러 biomarker들을 억제 또는 활성화하였다. 또한 UDCA의 BDC 세포의 EMT 억제효과는 ROS의 생성의 억제에 따른 PRX2와 STAT3 활성화를 방해함으로써 이루어졌을 가능성을 보여주었다. 따라서 UDCA는 단독으로 또는 기존 항암제와의 병합요법으로 BDC의 성장 및 침윤을 억제할 수 있는 항암제로서, 또는 보조치료제로서 기여할 수 있는 가능성을 확인하였다. 덧붙여, BDC 발생의 고위험군에서 UDCA를 사용한 적극적인 예방의 가능성도 있다고 생각한다.